
The clinical development of targeted therapeutics for myeloproliferative neoplasms (MPN) requires analytical precision to quantify true disease modification. Legacy analytical methods, such as bulk next-generation sequencing and multiparametric flow cytometry, suffer from inherent subclonal masking and force scientists to computationally infer the critical relationships between a cell's genotype and phenotype. Here we examine the somatic driver mutations underlying MPN pathogenesis and details how single-cell multiomics eliminates this reliance on inference by directly linking molecular profiles. Discover how simultaneous, cell-by-cell resolution of targeted DNA loci and surface proteins enables developers to rigorously characterize a novel therapy's mechanism of action, track the phylogenetic emergence of any subclonal mechanism of resistance, and establish definitive single-cell minimal residual disease (scMRD) endpoints to develop robust therapies to treat MPNs.
What are myeloproliferative neoplasms (MPNs) and what mutations cause the disease to form?
Myeloproliferative neoplasms (MPNs) are a group of Philadelphia chromosome-negative hematologic malignancies. They are characterized by the unregulated overproduction of myeloid cells across erythroid, myeloid, and megakaryocytic lineages, including erythrocytes, leukocytes, and megakaryocytes, within the bone marrow. MPNs form due to acquired somatic mutations in hematopoietic stem cells. These driver mutations cause hyperactivation of the Janus kinase/signal transducer and activator of transcription (JAK-STAT) signaling pathway.
This constitutive oncogenic signaling forces cellular proliferation, inhibits apoptosis, and alters the bone marrow microenvironment, leading to systemic inflammation and fibrosis. Patients diagnosed with myeloproliferative neoplasms face high risks of arterial and venous thromboembolic events, massive splenomegaly, and severe constitutional symptoms. Furthermore, MPNs carry a constant risk of leukemic transformation into secondary acute myeloid leukemia (sAML), an aggressive blast-phase disease with a highly unfavorable prognosis.
What are the different forms and subtypes of diseases categorized under MPN?
Clinical diagnostics categorize classical myeloproliferative neoplasms into three primary subtypes based on distinct morphological, clinical, and genotypic parameters: Polycythemia Vera (PV), Essential Thrombocythemia (ET), and Myelofibrosis (MF).
- Polycythemia Vera (PV): PV is defined by trilineage bone marrow hyperplasia (panmyelosis) that is heavily skewed toward the erythroid lineage. This results in an elevated red cell mass and high hematocrit. Over 99% of PV cases harbor JAK2 mutations, predominantly the V617F point mutation (~96%), with the remainder carrying rare JAK2 exon 12 alterations.
- Essential Thrombocythemia (ET): Characterized by marked, isolated thrombocytosis and bone marrow megakaryocytic hyperplasia without significant reticulin fibrosis. ET is generally the most indolent subtype. Its mutational profile is heterogeneous, involving JAK2, CALR, and MPL mutations.
- Myelofibrosis (MF): MF includes both primary myelofibrosis and secondary forms (post-PV or post-ET). It is the most aggressive MPN subtype, marked by atypical megakaryocyte proliferation, dense reticulin and collagen marrow fibrosis, massive splenomegaly, leukoerythroblastosis, and progressive cytopenias. MF shares the heterogeneous mutational profile of ET but carries a drastically reduced life expectancy and a higher risk of leukemic transformation.
Table 1: Subtype Myeloproliferative neoplasms (MPN) subtypes
| Clinical Parameter | Polycythemia Vera (PV) | Essential Thrombocythemia (ET) | Myelofibrosis (MF) |
|---|---|---|---|
Prevalence | 44-57 cases per 100,000 | 38-57 cases per 100,000 | 4-6 cases per 100,000 |
Median Age at Diagnosis | ~60 years | ~60 years | >65 years |
Blood Cell Abnormalities | High hematocrit; increased red cell mass | Elevated platelets; normal erythrocytes | Reduced erythrocytes; variable leukocytes |
Bone Marrow Pathology | Trilineage myeloproliferation | Increased megakaryocytes | Fibrosis, atypical megakaryocytes |
Prevalence of JAK2 Mutation | >99% | ~50% | ~50% |
What specific driver mutations cause MPNs?
Classical MPNs are initiated by somatic driver mutations in one of three genes, JAK2, CALR, or MPL, which are largely mutually exclusive with one another as primary oncogenic drivers.
- JAK2 Mutations: The JAK2 V617F mutation occurs in exon 14, altering the auto-inhibitory pseudokinase domain (JH2) of the Janus kinase 2 protein. This abrogates regulatory function, causing cytokine-independent activation of the adjacent kinase domain (JH1) and continuously phosphorylating downstream STAT proteins.
- CALR Mutations: Pathogenic CALR mutations are exclusively exon 9 insertions or deletions (such as the Type 1 52-base pair deletion or the Type 2 5-base pair insertion). This frameshift removes the endoplasmic reticulum retention signal (KDEL) and transforms the C-terminus into a highly basic, positively charged domain. The mutant CALR protein binds directly to MPL via the novel positively charged C-terminal domain, driving ligand-independent receptor activation and constitutive downstream JAK-STAT signaling.
- MPL Mutations: Alterations at codon 515 (e.g., W515L or W515K) induce a conformational shift in the juxtamembrane domain of the thrombopoietin receptor. This structural change mimics endogenous ligand binding, directly forcing receptor dimerization and independent JAK-STAT signaling.
A minority of ET and MF cases (~10–15%) are triple-negative. Some harbor mutations in non-canonical JAK-STAT regulators such as LNK/SH2B3 or CBL, while others remain genomically uncharacterized.
For developing a therapy to treat MPN, what are the current methods and their limitations?
Historically, MPN therapy development focused on symptom palliation and thrombotic risk mitigation rather than eradicating the underlying malignant clone. Standard cytoreductive therapies successfully control hematocrit levels but fail to alter the disease's natural history. Approved JAK1/2 inhibitors effectively manage inflammatory symptom burden and reduce spleen volume, but their lack of selectivity — inhibiting both wild-type and mutant JAK2, as well as JAK1 — frequently induces dose-limiting cytopenias and fails to deliver deep molecular remissions. When developing a therapy to treat MPN, researchers encounter severe technical limitations with conventional analytical methods such as bulk next-generation sequencing (NGS), flow cytometry, and the need to run multiple assays in different labs leading to inferred data and time wasted:
- Subclonal Masking: Bulk sequencing homogenizes nucleic acids from millions of diverse cells into a single, averaged dataset. In highly heterogeneous neoplasms like MPNs, this averaging masks the true subclonal architecture, obscuring rare, high-risk pre-leukemic subclones (such as TP53 or ASXL1 mutations) that drive the transformation to secondary AML.
- Zygosity Blindness: Bulk NGS cannot determine zygosity or mutational co-occurrence. It cannot distinguish if a JAK2 mutation and a secondary epigenetic mutation exist within the exact same malignant cell or in separate, competing clonal lineages.
- CHIP Confounding: Monitoring MPN therapy efficacy via bulk sequencing methods and flow cytometry is heavily confounded by Clonal Hematopoiesis of Indeterminate Potential (CHIP). Bulk assays and flow cytometry cannot differentiate between a benign, differentiated myeloid cell carrying a naturally accumulating CHIP mutation (such as DNMT3A or TET2) and a true pathogenic leukemic blast.
Why is there value in using single-cell multiomics in MPN therapy development?
To bypass the inherent limitations of these legacy methods, single-cell multiomics is the most effective analytical technology to implement in MPN drug development. By isolating individual cells to simultaneously analyze targeted DNA sequences, RNA transcripts, and cell-surface proteins, single-cell multiomics connects genotype to phenotype on a strict cell-by-cell basis.
- Precise Clonal Tracking: In MPNs, single-cell multiomics enables researchers to construct precise phylogenetic trees, tracing the temporal sequence of mutation acquisition and explicitly mapping the specific clonal hierarchies that drive disease progression.
- Single-Cell Minimal Residual Disease (scMRD): Single-cell multiomics redefines the criteria for clinical trial endpoints. Rather than relying on surrogate clinical markers, scientists utilize scMRD profiling to quantify true disease modification. By correlating immunophenotypic markers (such as CD34+ and CD117+) directly with somatic mutational profiles such as CALR or JAK2, an scMRD assay differentiates pathogenic mutant blasts from benign clonal hematopoiesis. This provides an ultra-sensitive mechanism to confirm the eradication of the mutant hematopoietic stem and progenitor cell (HSPC) pool.
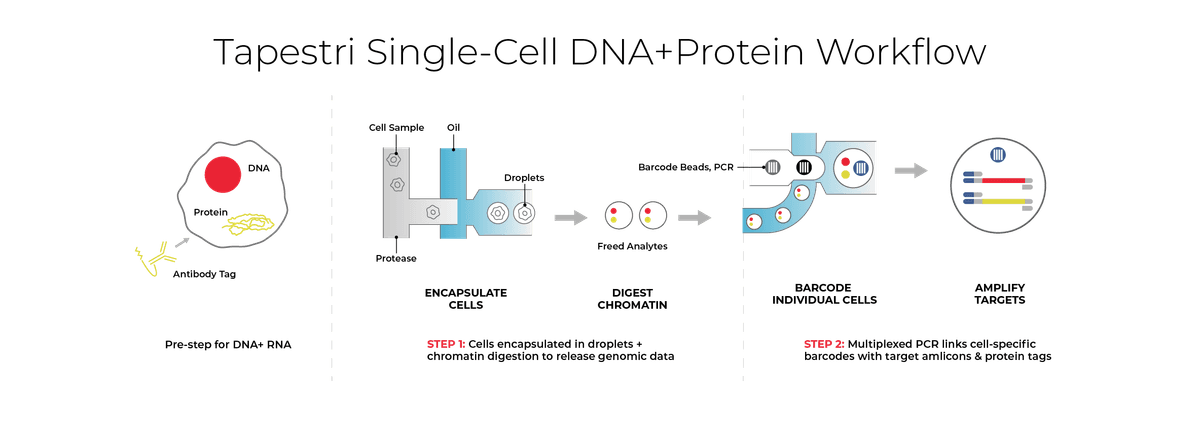
- Resolving Complex Target Genotypes: Specialized commercial workflows, such as the Mission Bio Tapestri platform, facilitate this single-cell resolution. The Tapestri workflow employs a two-step microfluidic droplet technology to encapsulate individual cells, tag targeted surface proteins with oligo-conjugated antibodies, and utilize targeted multiplex PCR to co-amplify the genetic loci and protein tags. This specifically resolves complex genetic alterations, such as CALR frameshifts, enabling researchers to accurately track the reduction of mutCALR positive HSPCs over whole blood variant allele frequency.
What current companies are developing drug molecules to treat MPN?
The pharmaceutical pipeline for MPNs is advancing rapidly, with Incyte leading the charge in precision therapeutics through their investigational molecule, INCA033989. This first-in-class, fully human monoclonal antibody is specifically designed to bind the pathogenic mutant calreticulin (mutCALR) protein and block its aberrant interaction with the thrombopoietin receptor (MPL). Unlike traditional therapies that broadly suppress the immune system or non-selectively inhibit kinase activity, INCA033989 physically binds to the mutant receptor complex on the surface of malignant cells, inducing internalization of the mutant receptor complex and suppressing aberrant signaling. This highly specific mechanism suppresses the oncogenic JAK-STAT signaling cascade while sparing healthy cells. In recent clinical data, INCA033989 demonstrated a favorable safety profile with no dose-limiting toxicities, rapidly normalizing platelet counts in essential thrombocythemia (ET) and producing clinically meaningful reductions in spleen volume and symptom burden in MF patients.
Discover how single-cell multiomics overcomes the analytical limits of legacy bulk sequencing. Watch this on-demand webinar showing how pre-clinical characterization of a monoclonal antibody to treat myeloproliferative neoplasms (MPN) using single-cell multiomics helped a company take their molecule into the clinic: Watch here.
To definitively prove that INCA033989 acts as a true disease-modifying agent rather than just a symptom palliative, Incyte utilized advanced single-cell multiomics to characterize the drug's effect at the cellular level. By analyzing patient biospecimens, researchers were able to track the exact genotypic and immunophenotypic changes within the hematopoietic stem and progenitor cell (HSPC) compartment. This high-resolution single-cell tracking validated that INCA033989 selectively eliminates the mutant CALR+ HSPC populations while promoting the restoration of normal, wild-type hematopoiesis. The success of this single-cell monitoring approach demonstrated the potential of single-cell multiomics to validate disease-modifying activity in a targeted therapy setting. This has coincided with growing interest across the MPN field in mutation-specific therapeutic strategies. Multiple biotechnology developers are now pursuing precision therapies targeting mutCALR and JAK2 V617F, the dominant driver mutations across MPN subtypes. Single-cell multiomics is increasingly being leveraged across these programs to validate the therapeutic activity of these molecules at the clonal level.
What is the clinical utility of single-cell multiomics for MPN?
The clinical utility of single-cell multiomics in MPN precision medicine is rooted in the fundamental inadequacy of legacy analytical methods to accurately measure true MPN disease mutations and their impact on disease progression. Traditional diagnostic and monitoring approaches rely heavily on bulk next-generation sequencing (NGS), RNA-sequencing (RNA-seq), and multiparametric flow cytometry. However, these legacy assays are no longer sufficient for evaluating next-generation therapies. Bulk sequencing inherently averages data across millions of cells, completely masking the critical subclonal architecture, mutational co-occurrence, and zygosity of the mutant MPN clones. Additionally, traditional bulk RNA-sequencing is not enough of a targeted approach to truly address a therapy's mechanism of action (MOA) or accurately trace the differentiation of leukemic blasts. Furthermore, running separate, parallel assays such as bulk DNA sequencing for genotype and a distinct flow cytometry run for immunophenotype forces scientists to computationally infer the relationship between a cell's mutations and its physical state. This indirect inference frequently results in false positives, such as mistaking benign clonal hematopoiesis for residual disease, as well as false negatives, where rare, therapy-resistant mutant subclones are missed entirely.
To rigorously prove that a targeted drug molecule successfully eradicates the malignant stem cell niche without inducing unwanted cellular responses, scientists must directly link genotype to phenotype. Depending on the specific modality of the therapy being developed, drug developers need to simultaneously analyze targeted DNA mutations alongside cell-surface protein expression, or integrate DNA profiling with targeted RNA transcription on a strict cell-by-cell basis like what the Tapestri® instrument can provide. This single-cell multiomic resolution ensures that scientists can explicitly link genotypic alterations, like the clearance of pathogenic JAK2 or CALR clones, directly with phenotypic states, allowing them to confidently differentiate true malignant leukemic blasts from benign cells and track the precise emergence of therapy-resistant subclones.
Because every targeted therapy operates via a unique mechanism of action, out-of-the-box analytical methods frequently fall short in characterizing a drug therapy during development. There is immense value in building custom, fit-for-purpose assays that are specifically engineered to meet the exact analytical needs of a particular MPN clinical program. By utilizing customized single-cell multiomic assays tailored to simultaneously measure specific DNA loci and the corresponding protein or transcriptomic markers, pharmaceutical developers and biotech companies can characterize their novel therapies effectively and comprehensively. This bespoke, high-resolution approach eliminates the blind spots of legacy assays, easing the path to regulatory approval and allowing effective, life-saving therapies to reach MPN patients.
To learn more about implementing an MPN custom single-cell multiomic assay for your therapy development pipeline, reach out to the experts or set up a call today with a specialist.
References:
- Mehta, J. "Epidemiology of Myeloproliferative Neoplasms in the United States." Leukemia & Lymphoma, vol. 55, no. 3, 2014, pp. 595-600.
- Stein, B. L. "Polycythemia Vera: An Appraisal of the Biology and Management 10 Years After the Discovery of JAK2 V617F." Journal of Clinical Oncology, vol. 33, no. 33, 2015, pp. 3953-3960.
- Johansson, P. "Epidemiology of the Myeloproliferative Disorders Polycythemia Vera and Essential Thrombocythemia." Seminars in Thrombosis and Hemostasis, vol. 32, no. 2, 2006, pp. 171-173.
- Girodon, François. "Significant Increase in the Apparent Incidence of Essential Thrombocythemia Related to New WHO Diagnostic Criteria: A Population-Based Study." Haematologica, vol. 94, no. 6, 2009, pp. 865–869.
- Gangat, N. "DIPSS Plus: A Refined Dynamic International Prognostic Scoring System for Primary Myelofibrosis That Incorporates Prognostic Information from Karyotype, Platelet Count, and Transfusion Status." Journal of Clinical Oncology, vol. 29, no. 4, 2011, pp. 392-397.
- Tefferi, A. "Survival and Prognosis Among 1545 Patients with Contemporary Polycythemia Vera: An International Study." Leukemia, vol. 27, no. 9, 2013, pp. 1874-1881.
- Klampfl, Thorsten. "Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms." New England Journal of Medicine, vol. 369, no. 25, 2013, pp. 2379–2390.
- Arber, Daniel A. "The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia." Blood, vol. 127, no. 20, 2016, pp. 2391–2405.
- Pardanani, A. "Prevalence and Clinicopathologic Correlates of JAK2 Exon 12 Mutations in JAK2V617F-Negative Polycythemia Vera." Leukemia, vol. 21, no. 9, 2007, pp. 1960-1963.
- Cervantes, F. "Improving Survival Trends in Primary Myelofibrosis: An International Study." Journal of Clinical Oncology, vol. 30, no. 24, 2012, pp. 2981-2987.
- Szuber, N. "3023 Mayo Clinic Patients with Myeloproliferative Neoplasms: Risk-Stratified Comparison of Survival and Outcomes Data Among Disease Subgroups." Mayo Clinic Proceedings, vol. 94, no. 4, 2019, pp. 599-610.
- Barbui, T. "Philadelphia-Negative Classical Myeloproliferative Neoplasms: Critical Concepts and Management Recommendations from European LeukemiaNet." Journal of Clinical Oncology, vol. 29, no. 6, 2011, pp. 761-770.



