What is acute myeloid leukemia?
Acute myeloid leukemia (AML) is an aggressive cancer of the blood and bone marrow. It begins when immature myeloid cells, the precursors that normally mature into red cells, platelets, and infection-fighting white cells, acquire mutations that lock them in a rapidly dividing, immature state. These cells, called blasts, crowd out healthy blood production and drive the disease.
AML is one of the most common leukemias and one of the most genetically diverse. Two patients can carry entirely different driver mutations, and within a single patient the disease is rarely uniform; it is a mosaic of related but genetically distinct clones. That genetic complexity is what makes AML so difficult to treat, and to measure, and it shapes everything that follows about measurable residual disease (MRD).
Relapse Remains the Definitive Barrier to AML Survival
Most patients with AML reach complete remission after induction. Yet relapse, not initial failure, remains the leading cause of death. Many patients in morphologic remission still harbor disease beneath standard detection, which can be a result of current standard of care (SoC) method limitations.
The leukemic cells that persist below morphologic detection are one of the strongest independent predictors of relapse and survival in AML. MRD has become a cornerstone of AML risk-adapted therapy intervention. This molecular insight directly informs decisions regarding transplant stratification, consolidation intensity, and early intervention prior to morphological relapse.
However, an MRD assessment is only as predictive as the clonal complexity it can resolve. In AML, that underlying architecture is exceptionally heterogeneous and dynamic.
Why measuring MRD in AML is uniquely difficult
AML is not one disease in one cell. It's a shifting population of genetically distinct clones, which is exactly what makes its residual disease so hard to measure.
Unlike chronic myeloid leukemia having the BCR-ABL fusion marker to identify the disease, AML has no marker present in every patient. MRD must be tracked across a patient-specific set of lesions.
Clonal heterogeneity
A leukemia is a mix of related clones with different mutation combinations. The clone that drives relapse is often not the dominant one at diagnosis.
CHIP confounds molecular MRD
Clonal hematopoiesis of indeterminate potential (CHIP) mutations can persist after therapy, confounding bulk DNA assays by mimicking residual leukemia.
Lineage plasticity
Under therapeutic pressure, cells shift differentiation state and surface phenotype, evading assays that assume a fixed immunophenotype.
Persistent pre-leukemic clones
Ancestral clones can survive therapy without being the cells that cause relapse, blurring what an MRD-"positive" result means.
No universal marker
Lacking a universal biomarker, AML requires a personalized panel for cellular or genetic MRD tracking.
The leukemic stem-cell problem
The cells most responsible for relapse are rare, quiescent, and phenotypically elusive, easy for population-level methods to miss entirely.
Critical limitations of current standard of care AML MRD detection methods
While current SoC MRD methods provide vital clinical utility, each ultimately faces fundamental technological limitations.
Multiparameter flow cytometry
Fast and widely available; reads the surface phenotype of thousands of cells.
Structural Limitation: operator-dependent, vulnerable to therapy-induced phenotype shift, and blind to genotype: it sees surface markers, not the mutations underneath.
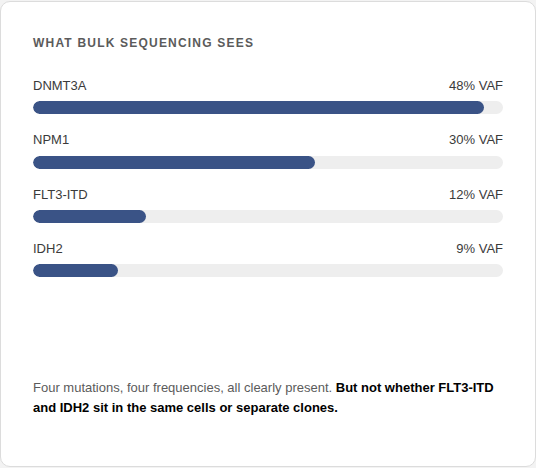
Bulk next-generation sequencing (NGS)
Sensitive to many mutations at once across the genome of a sample.
Structural Limitation: reports population averages. A variant allele frequency tells you a mutation is present, not whether two mutations share a cell, and not whether a signal is leukemia or CHIP.
qPCR / ddPCR for fusion transcripts
Good sensitivity for tracking defined molecular targets, such as NPM1 mutations and select fusion transcripts.
Structural Limitation: restricted to known, pre-defined targets. Most AML cases lack the universal or recurrent driver mutations necessary for this type of targeted assay.
These assays cannot link genotype to phenotype. By failing to simultaneously measure mutation co-occurrence and cell-surface immunophenotypes, legacy standard of care methods merely assess isolated parts rather than the underlying clonal architecture.
SoC vs single-cell readout comparison for AML MRD
MRD Case | Cells Involved | Single-Cell Multiomics Readout | Flow Cytometry | Bulk NGS | Standard of Care Readout |
|---|---|---|---|---|---|
True Negative | Healthy mature and stem cells | Correct | X | X | Correct |
False Positive | Clonal hematopoiesis of indeterminate potential (CHIP) | Correct | X | ✔️ | Ambiguous |
False Positive | Aberrant regenerating stem cell | Correct | ✔️ | X | Ambiguous |
False Negative | Rare subclones (<0.1% burden) | Correct | X | X | Not Detectable |
True Positive | Classic AML blast | Correct | ✔️ | ✔️ | Correct |
How Single-Cell Multiomics Overcomes Current AML MRD Limitations
Rather than tracking isolated genetic mutations and cell-surface immunophenotypes independently, single-cell multiomics pairs genotype with phenotype within individual cells. This shifts MRD evaluation from disconnected readouts that obscure subclonal diversity to the simultaneous resolution of rare residual clones and cellular architecture that is critical for AML.
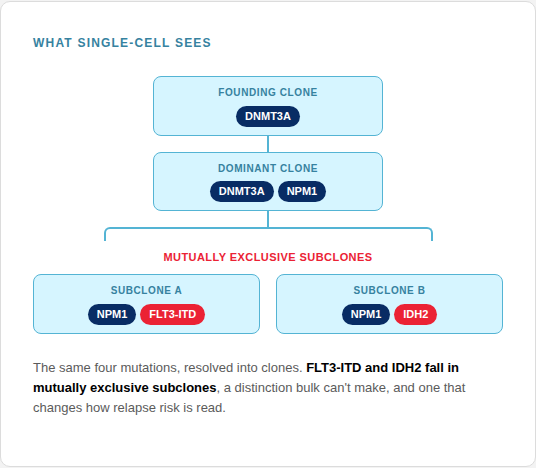
Co-occurrence & clonal architecture
Genotyping thousands of single cells uncovers precise mutational co-occurrence, allowing the direct reconstruction of the true clonal hierarchy rather than inferring it from averages.
Rare-clone detection
Because each cell is its own measurement, a small but dangerous relapse-initiating clone shows up even when invisible in a population average.
Zygosity, cell by cell
Measuring individual cellular zygosity resolves true clonal dominance and loss-of-heterozygosity, mapping critical clonal architectures missed by bulk VAF measurements.
Genotype + immunophenotype together
Co-measuring a cell's DNA and surface markers explicitly links mutations to individual immunophenotypes. This distinguishes benign CHIP from malignant leukemia and traces the underlying genotype directly to the primitive stem-cell compartment.
Clinical and translational applications of single-cell multiomics in AML MRD detection
The clinical utility of single-cell multiomics has the ability to map therapeutic clonal evolution, predict resistance to therapies sooner, and define high-resolution clinical trial endpoints.
Clonal-evolution tracking under therapy
Watching which clones contract, persist, or expand across treatment, rather than seeing only a net signal rise or fall.
Guiding therapy strategies
Characterizing single-cell mutation co-occurrence and surface biomarkers like CXCR4 can aid patient stratification, distinguishing therapeutic responders from nonresponders.
Detect resistant clones before relapse
Identifying therapeutically resistant mutant clones while intervention is still possible to mitigate relapse.
Sharper clinical-trial endpoints
Single-cell MRD readouts enhance AML trial endpoints, providing precise response definitions that could enhance the evaluation of overall and relapse-free survival.
Key publications evaluating single-cell AML MRD detection
Peer-reviewed evidence demonstrating how single-cell resolution refines AML trial endpoints, characterization of response, and survival metrics for acute myeloid leukemia.
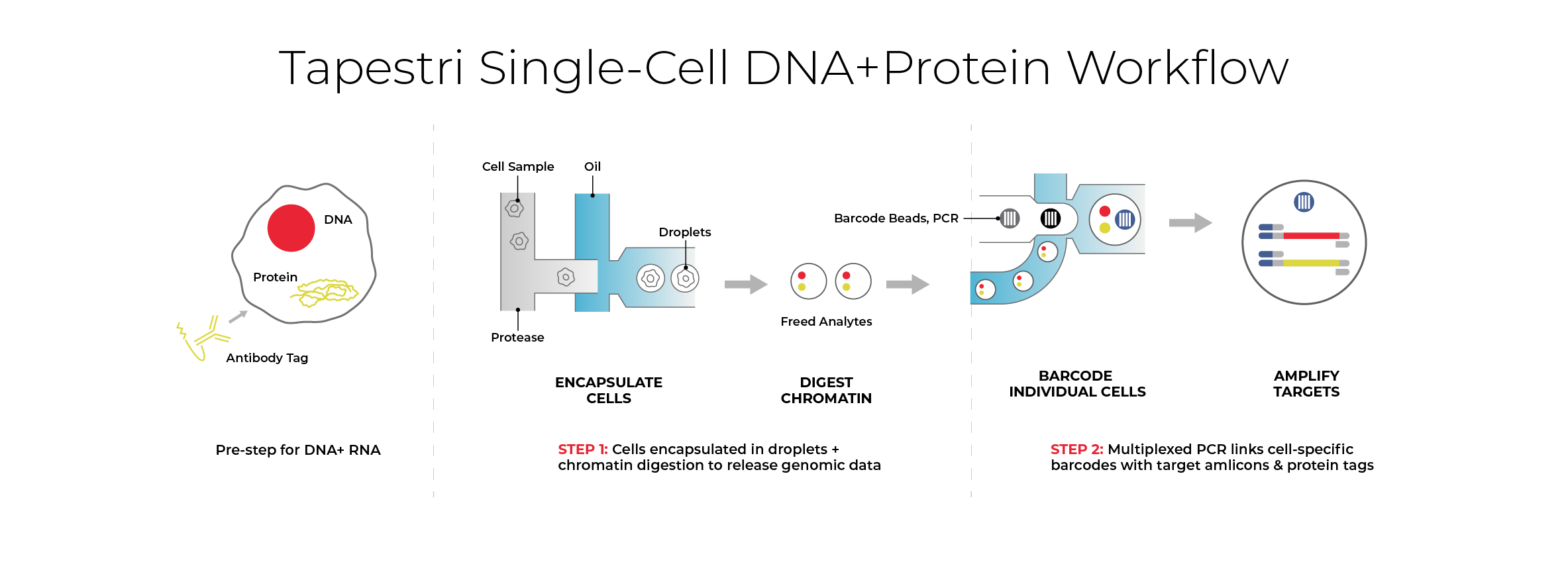
How Mission Bio's Tapestri® Platform enables single-cell MRD detection
The Tapestri Platform is a microfluidic system designed to co-measure DNA mutations and cell-surface protein expression simultaneously within individual cells. By deploying the Tapestri Single-cell MRD (scMRD) AML Assay, which pairs high-sensitivity genotypic sequencing with immunophenotypic profiling, scientists can track complex clonal evolution and distinguish true residual leukemia from pre-leukemic clones. This dual-modality approach also establishes a clear path toward clinical utility, offering the analytical resolution necessary to support guided therapeutic strategies and precise endpoint tracking.
Built for seamless scalability and tech transfer, this workflow can be implemented directly within your own laboratory setting, transitioned to preferred CRO and CDMO partners, or executed as a managed project through Mission Bio's Assay Services.
For research use only. Not for use in diagnostic purposes.