
As CRISPR-based approaches continue to be implemented across applications such as disease modeling, functional genomic, screening, and cell & gene therapy, the ability to resolve editing outcomes at the single-cell level has become a fundamental requirement. Each application demands not only confirmation that an edit occurred, but a precise understanding of its consequences such as how it alters gene expression, protein output, genome integrity, and safety within a heterogeneous population. Single-cell multiomics meets this requirement by integrating genomic, transcriptomic, and proteomic insights within the same cell, integrating siloed molecular methods into a unified single assay.
Single-Cell Sequencing Vol 2: The Essentials.
Single-Cell 101, Volume 2: The Essentials takes you beyond the basics, with new content on cell and gene therapy (CGT) and single-cell RNA analysis.
How Does Single-Cell Multiomics Help CRISPR-generated Disease Model?
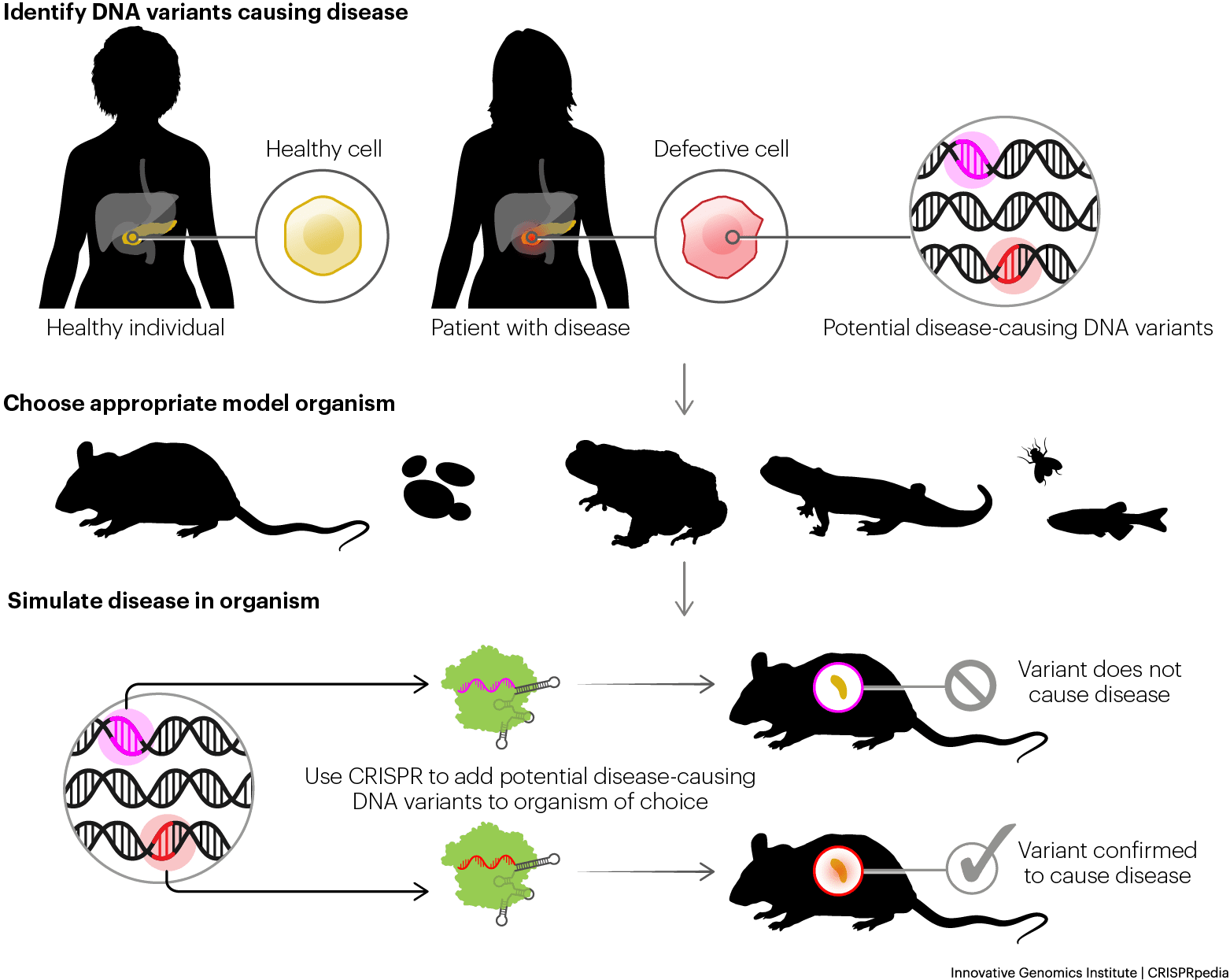
Disease modeling relies on CRISPR to engineer precise, disease-associated mutations into immortalized cell lines, human organoids, and animal models including mice and zebrafish, to study pathology and identify therapeutic vulnerabilities (figure 1). However, the biological fidelity of these models is only as informative as the analytical methods applied to characterize them. Confirming that an intended mutation was introduced at the correct locus, in the correct zygosity, and without co-occurring off-target alterations is a prerequisite for drawing valid conclusions from any disease model which bulk methods fundamentally cannot satisfy. Undetected off-target editing events that co-exist within the model system introduce confounding phenotypic readouts that can misrepresent disease biology, producing false conclusions about the role of the intended genetic driver.
Single-cell multiomics addresses this in disease models to ensure complete characterization. On-target indels and base edits are confirmed at the single-cell level while off-target events are simultaneously flagged, ensuring that observed phenotypes can be attributed to the intended perturbation rather than an uncharacterized secondary genetic edit. Allelic resolution further confirms mono- versus bi-allelic editing status in each individual cell, which is particularly critical for recessive disease models where homozygous knockout is required for phenotype expression; a heterozygous cell population masquerading as a complete knockout represents a fundamental model integrity failure. Multiplex CRISPR gene edit co-occurrence is similarly should be resolved at the per-cell level, confirming that all intended edits are present together within the same cell rather than distributed across distinct subpopulations.
Critically, confirmed editing status is directly linked to its transcriptional consequence within the same cell. This enables researchers to identify not only that a targeted knockout or knock-in is present, but that it produces the expected downstream gene expression change and to flag cells where an edit is confirmed at the DNA level but the transcriptional effect is absent or aberrant, a finding that can profoundly impact model interpretation and validity. In animal models, mosaic editing outcomes where distinct cell populations within the same tissue carry different CRISPR gene edit configurations are resolved with the same per-cell precision using single-cell multiomics. Xenograft models benefit further from using single-cell multiomic assays, as the clonal dynamics of engrafted human tumor cells can be tracked against a background of host-derived cells, with editing status, transcriptional state, and surface protein expression co-measured within the same cell to confirm that a targeted genetic alteration drives the expected shift in cell identity or marker expression within the model system. This level of concurrent genotypic to phenotypic understanding further validates disease models for understanding pathology and evaluating therapeutic intervention.
Benefits of Using Single-Cell Multiomics For Organoids Applications
Organoids are three-dimensional in vitro cellular assemblies derived from pluripotent stem cells or tissue-resident progenitors that self-organize to recapitulate the microanatomy, cellular heterogeneity, and functional dynamics of primary organs. While these models accurately reflect physiological tissue architecture, their inherent biological complexity complicates the evaluation of CRISPR editing outcomes. During the structural differentiation of stem cell-derived organoids, gene editing efficiency and mutation profiles vary significantly across emerging cell lineages. Standard bulk sequencing requires tissue homogenization, obscuring this lineage-specific cellular context. Consequently, bulk variant allele frequency data cannot confirm whether a genetic edit successfully penetrated the target cell lineage or if the signal is derived from highly edited, non-target cell types.
Single-cell multiomics resolves this complexity following organoid dissociation. By profiling individual cells, this approach identifies distinct cellular lineages via targeted transcriptomic or immunophenotypic signatures while simultaneously confirming genomic editing status within those exact subpopulations. This enables the precise verification of on-target indels, zygosity, and off-target events within the specific cell types driving disease pathology. By directly linking the confirmed genotype to its downstream transcriptional consequence in the same cell, researchers can evaluate how targeted CRISPR gene edits or multiplexed gene edits impact lineage-specific differentiation and cellular states without the masking effects of population averages some current methods leave you with. Ultimately, integrating single-cell multiomics into organoid development validates the biological fidelity of the model. By definitively confirming that observed pathological phenotypes are driven exclusively by the intended genetic alterations rather than off-target artifacts or unedited background cells, researchers ensure the strict reliability of their organoids for downstream mechanistic discovery and therapeutic screening.
In Vivo Animal Models Validation With Single-Cell Multiomics
In vivo animal models, such as genetically engineered mice or zebrafish, provide a systemic physiological context to study human genetic diseases and evaluate therapeutic interventions. However, generating these models via embryonic microinjection or systemic delivery of CRISPR components like electroporation and lipofection frequently results in highly heterogeneous editing outcomes. For example, embryonic CRISPR gene editing often yields heterogeneity, creating a founder generation with complex mutational profiles. In cases like zebrafish, you would need to create subsequent crossbreed generations to eventually generate the desired disease model. To ensure the desired genetic edit is presented on all alleles in the daughter zebrafish population, single-cell multiomics can play a critical role in this assessment. Furthermore, systemic delivery introduces critical biodistribution challenges, as delivery vehicles like lipid nanoparticles or viral vectors often exhibit unequal tissue tropism, accumulating heavily in organs like the liver while failing to reach intended extrahepatic targets. Standard bulk sequencing of tissue biopsies homogenizes this cellular diversity. Consequently, bulk variant allele frequencies fail to distinguish between a uniformly edited tissue and a mosaic tissue, and they completely obscure the exact cellular biodistribution of a CRISPR gene edit among the diverse cell lineages comprising a complex organ within the disease model.
Single-cell multiomics resolves this genetic ambiguity and provides high-resolution biodistribution profiling following tissue dissociation. By analyzing individual cells harvested from multiple anatomical sites, researchers can definitively quantify the precise zygosity and proportion of edited cells distributed across the organism. This approach maps the exact cellular biodistribution of the CRISPR gene edits, utilizing simultaneous transcriptomic or immunophenotypic profiling to identify precisely which cell lineages within a tissue successfully integrated the modification. By directly linking the confirmed genotype to its downstream transcriptional consequence in the exact same cell, researchers can evaluate the true functional impact of the intended gene edit in vivo. Ultimately, applying single-cell multiomics to animal models guarantees the genetic fidelity and spatial characterization of the organism. By definitively confirming that the intended mutation is present in the correct allelic state, properly localized to the target cell types, and drives the expected physiological phenotype, researchers ensure that costly, long-term pharmacological studies are not confounded by undetected mosaicism, skewed biodistribution, or hidden off-target artifacts.
Using Single-Cell Multiomics In Functional Genomics and Screening
Functional genomics and screening aims to systematically uncover the roles of genes and non-coding regulatory elements within complex biological networks. High-throughput pooled CRISPR screens achieve this by disrupting thousands of genetic targets in parallel and observing the resulting cellular responses across a heterogeneous population. Furthermore, CRISPR screens are instrumental in uncovering the mechanism of action (MoA) and mechanisms of resistance (MoR) of targeted therapeutics, providing actionable data to de-risk preclinical development and optimize future clinical trials. However, traditional pooled screening approaches are fundamentally constrained by low-content readouts measuring binary outcomes such as cell survival or single marker expression that provide no mechanistic insight into why a CRISPR gene edit produced a given phenotype. Critically, the inability to confirm whether a guide RNA delivered to a cell produced a functional edit leaves screen hits highly vulnerable to both false positives, driven by incomplete editing, cellular mosaicism, or in-frame indels that fail to trigger nonsense-mediated decay, and false negatives, where true biological effects are diluted or masked by the presence of unedited or partially edited cells within the same population.
Single-cell multiomics addresses these constraints within a single workflow. Indel frequency, on-target & off-target assessment, and edit rate are quantified across all library targets simultaneously, enabling direct ranking of editing efficiency and flagging of off-target-enriched hits before committing resources to functional follow-up. Allelic states and multiplex edit co-occurrences are resolved at the per-cell level, deconvoluting incomplete editing events and ensuring that screen hits are driven by complete target disruption rather than partial or heterozygous edits that may produce attenuated or compensatory phenotypes. By directly linking each confirmed CRISPR gene edit to its transcriptional response within the same cell, true on-target regulatory effects are confidently distinguished from cells lacking edits or exhibiting compensatory transcriptional states eliminating a primary source of both false positive and false negative hit calling.
The resolution of single-cell multiomics further extends to the proteome, enabling screen hits that drive changes in surface protein expression to be identified by directly linking genome edits to immunophenotype within individual cells. This allows prioritization of candidates with discrete, cell surface-measurable phenotypes without requiring cell sorting or split-sample workflows. During hit validation, candidate genes identified in a primary screen must be rigorously evaluated to establish true causality between a confirmed genomic edit and the observed biological effect. By simultaneously verifying edit completeness at the DNA level and measuring the intended functional consequence at the RNA and/or protein level within the same cell, off-target confounds are resolved and only mechanistically validated targets advance in the drug discovery pipeline while also mitigating the presence of false-positive and false-negatives.
Single-Cell Multiomics Provides Safety and Genotoxicity Assessments Need in Cell and Gene Therapy Development
The development of CRISPR-based cell and gene therapies encompasses both ex vivo engineered cellular products, such as autologous and allogeneic CAR-T cells or hematopoietic stem cells, and in vivo modalities utilizing viral vectors or lipid nanoparticles to directly edit patient tissues inside their body. Regardless of the delivery mechanism or how the therapy is manufactured, these advanced therapies require rigorous quality control to ensure precise on-target efficacy, compatibility during manufacturing, and absolute patient safety. Regulatory agencies are continuously changing guidelines for cell & gene therapies, requiring detailed characterization of all gene-edited therapeutic products. Consequently, the analytical standards applied to these therapies continue to escalate. Conventional methodologies that report population-averaged signals or rely on multiple fragmented workflows to evaluate Critical Quality Attributes (CQAs) are now fundamentally insufficient. These legacy approaches fail to provide the granular, cell-by-cell evidence that regulators are wanting to see for robust Investigational New Drug (IND) and Biologics License Application (BLA) submissions.
Single-cell multiomics used in cell and gene therapy development addresses this by consolidating the CQAs of a gene-edited therapeutic into a single, fit-for-purpose workflow. On- and off-target editing outcomes are quantified simultaneously within the same cell, enabling developers to answer the question regulators are increasingly asking: do cells harboring dangerous off-target events also carry the intended therapeutic modification? This co-occurrence understanding replaces the ambiguity of decoupled bulk datasets with definitive, per-cell evidence that the tissue sample or cell therapy product is free of compounded genomic safety risks. Additionally zygosity is resolved at the single-cell level, confirming whether edits are heterozygous or homozygous across both alleles , a distinction that directly impacts potency modeling, genotoxicity assessment, and the definition of an effective therapeutic dose that legacy analytical assays lack assessing. For multiplex CRISPR gene editing strategies, co-occurrence analysis verifies that all intended edits are present together within the same cell, ensuring the final product is not a mixed population of partially edited cells but a compositionally defined therapeutic.
Learn how single-cell DNA sequencing comprehensively validated the safety of a new CRISPR gene therapy for Hyper-IgM 1. In the recording, see how single-cell DNA sequencing helped rule out off-target effects and supported first-in-human trials.
Using single-cell multiomics, vector copy number (VCN) can be quantified at single-cell resolution, revealing the true distribution of transgene integration across the product population rather than a misleading average. When integrated with targeted gene expression profiling in the same cell, this distinguishes transcriptionally active, therapeutically functional cells from those harboring silent vector copies providing a more rigorous metric for functional titer and potency that supports rational optimization of transduction conditions and process parameters. Importantly, chromosomal aberrations and large structural variants that standard indel-focused assays frequently miss are simultaneously assessed, ensuring genome integrity is characterized at the resolution required to detect rare but potentially hazardous rearrangements before they reach the clinic.
For in vivo gene therapies, biodistribution studies introduce an additional layer of analytical complexity that legacy methods are fundamentally ill-equipped to resolve. Following administration of an in vivo gene-edited therapeutic, the edited cell population distributes across tissues and organs where it encounters distinct microenvironments that can differentially influence engraftment, expansion, and therapeutic activity. Bulk analysis of recovered cells from these tissues produces population-averaged signals that obscure the true composition of the engrafted population masking the proportion of cells that retain the intended edit, have undergone clonal expansion, or have acquired secondary genomic alterations in vivo. Single-cell multiomics resolves this by simultaneously characterizing editing status, transcriptional state, and immunophenotype in each recovered cell, enabling precise tracking of the therapeutic population across tissue compartments. Clonal dynamics are resolved at single-cell resolution, identifying whether specific edited subclones are preferentially expanding or contracting within a given tissue context, a critical safety consideration for therapies where clonal dominance driven by off-target editing represents a genotoxic risk. This per-cell resolution across biodistribution samples provides the definitive evidence needed to confirm that the therapeutic product maintains its intended genomic and functional integrity at the site of action.
Crucially, single-cell multiomics directly anchors these complex genomic observations to their downstream functional consequences. By simultaneously correlating a confirmed genotype with targeted transcriptional modulation and discrete immunophenotypic shifts, developers can definitively validate that cells harboring the intended edit combination also exhibit the requisite therapeutic potency profile. This consolidated multiparametric readout eliminates the inferential gap inherent to decoupled legacy assays. Ultimately, single-cell multiomics delivers the determinative genotype-to-phenotype evidence needed to rigorously characterize CQAs and establish manufacturing comparability, thereby de-risking regulatory submissions and obviating the need for legacy, fragmented analytical workflows.
Learn how single-cell DNA sequencing provides the comprehensive insights essential for any CRISPR experiment, including zygosity, co-occurrence, and critical safety assessments such as off-target effects and translocations.
Single-Cell Multiomic Platform For CRISPR Gene Editing Application
The continued use of CRISPR gene editing across disease modeling, functional genomics and screening, and cell & gene therapy necessitates a fundamental shift in how the CRISPR gene editing is characterized to ensure success in experiments and therapy development. Single-cell multiomics provides the indispensable data required to overcome the limitations of legacy assays, enabling the precise quantification of zygosity, identify on-target and off-targets, evaluate multiplex edit co-occurrence, transcriptional states, immunophenotypical readouts and complex biodistribution within complex organisms. Furthermore, by simultaneously capturing genomic and downstream functional data within the exact same cell, this methodology establishes the definitive genotype-to-phenotype linkage critical for mechanistic validation and clinical safety. Mission Bio provides the targeted single-cell multiomic solutions required to navigate these complex analytical demands. Through the multiparametric Tapestri platform and specialized assay development services, Mission Bio enables researchers and therapeutic developers to consolidate fragmented quality control matrices into a single, high-resolution workflow, delivering the determinative cellular evidence needed to de-risk therapeutic pipelines and provide definitive data needed in manuscript or grant proposals.
Contact our team today to discover how Mission Bio's single-cell multiomic solutions and custom assay development services can comprehensively de-risk your CRISPR pipeline and accelerate your clinical timelines
