
The adaptation of clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins has transformed biological research by enabling the precise manipulation of genomic function. This gene editing technology is now being utilized in a variety of applications such as the generation of accurate disease models, the execution of complex functional screens, and the development of curative cell and gene therapies. Despite the precision of CRISPR gene editing, accurately characterizing the molecular outcomes of a genetic perturbation requires overcoming substantial analytical barriers. Implementing rigorous CRISPR analyses is mandatory for evaluating the genotoxicity, efficacy, and complex cellular responses associated with gene editing. Achieving this level of evaluation necessitates an exhaustive assessment of genomic integrity, transcriptional modulation, and phenotypic shifts. The transition from traditional bulk sequencing methods to single-cell multiomics provides the critical framework required for detailed CRISPR analysis, allowing scientists to directly link their CRISPR editing to specific genotypes and their resulting phenotypes without the confounding variability when using fragmented analysis methods that waste time and resources.
Technical Challenges in Gene Editing Analysis
One primary concern in any CRISPR gene editing application is the occurrence of off-target effects. Off-target gene editing happens when the CRISPR effector (e.g. Cas9) induces double-strand breaks at unintended genomic loci. This is primarily driven by tolerance of the Cas9–sgRNA complex to base pair mismatches in the protospacer, particularly in the PAM-distal region, where mismatches are more readily accommodated than in the critical PAM-proximal seed sequence. The deposition of these unintended alterations poses severe genotoxic risks, potentially disrupting essential genetic regulatory elements or the dysregulated expression of tumor suppressor genes. Consequently, robust CRISPR analysis workflows must accurately map these off-target cleavage events across the entire genome to ensure cellular safety and mitigate genotoxicity.
Mosaicism presents another significant challenge, arising when a CRISPR gene editing event does not occur uniformly across all cells within a target population or developing embryo resulting in a heterogeneous sample composed of gene edited and wildtype cells. For example, injecting Cas9 into zygotes frequently results in varying mutation profiles among the dividing daughter cells, leading to a complex mixture of wild-type, monoallelic, and biallelic mutant cells. This cellular heterogeneity is averaged in bulk sequencing readouts, making it highly difficult to establish stable, uniformly edited lineages for downstream research or therapeutic use.
This means determining the exact zygosity and tracking the co-occurrence of mutations is critical for understanding the functional impact of an edit. Bulk sequencing cannot definitively distinguish whether a fifty percent variant allele frequency represents heterozygous monoallelic edits present in all cells or homozygous biallelic edits present in only half of the cell population. Furthermore, multiplexed CRISPR editing aims to alter multiple genes simultaneously. Analyzing the co-occurrence of these multiple intended edits within a single cell is analytically demanding but absolutely necessary for evaluating pathway interactions and combinatorial drug targets.
Beyond localized insertions and deletions, CRISPR nuclease induced double-strand breaks frequently trigger massive chromosomal aberrations during the DNA repair process. Cellular repair mechanisms, including non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ), can generate megabase-scale deletions, translocations, inversions, and chromothripsis, also known as chromosomal shattering. Copy-neutral loss of heterozygosity is also frequently observed, which can unmask recessive pathogenic variants including inactivating mutations in tumor suppressor genes creating significant oncogenic risk. Standard short-read amplicon sequencing often misses these large structural variants entirely, creating a dangerous blind spot in conventional CRISPR gene editing analysis.
A core objective of functional genomics is definitively linking specific genetic perturbations to their downstream transcriptional readouts. However, significant technical barriers exist when attempting to correlate CRISPR-induced genomic edits with their resulting expression profiles. For instance, if a targeted CRISPR edit alters a specific transcription factor, accurately capturing the subsequent cascade of differential gene expression requires the simultaneous co-detection of the precise genomic sequence and the resulting mRNA transcripts within the exact same cell.
Similarly, associating CRISPR edits with the phenotype means linking specific genomic alterations directly to protein expression such as cell surface markers. Current analytical methods inherently struggle with this genotype-to-phenotype correlation because they rely on fragmented bulk assays that process DNA and Protein readouts in separate sample aliquots. This decoupled approach destroys the critical biological context of the individual cell, forcing developers to rely on statistical inferences about population averages rather than providing the direct, definitive proof required to confirm that a CRISPR gene edit successfully eliminated the target protein on the cell surface.
For in vivo cell and gene therapy applications, the biodistribution or delivery of CRISPR components presents significant logistical and analytical challenges. Viral vectors, such as adeno-associated viruses (AAV) and lentivirus, face packaging capacity constraints and potential immunogenicity upon administration. Non-viral vectors, including lipid nanoparticles, predominantly traffic to the liver, complicating the targeting of extrahepatic tissues. Tracking precise spatial localization and editing efficiency within specific target organs remains a critical hurdle for clinical CRISPR analysis.
Finally, allele dropout is a critical technical failure in polymerase chain reaction and next-generation sequencing workflows used for gene editing validation. When CRISPR induces large structural deletions or rearrangements that disrupt primer binding sites or amplicon integrity, the modified allele fails to amplify efficiently. Consequently, the sequencing assay only detects the remaining wild-type allele, resulting in a false-negative readout for the editing event. Overcoming allele dropout requires methodologies capable of detecting copy number variations independently of targeted short amplicons.
Standard Assays for CRISPR Analysis and Their Limitations
Evaluating gene editing outcomes requires distinct analytical approaches at the genomic, transcriptomic, and proteomic levels. While current standard assays provide valuable baseline data, their inherent technical limitations necessitate careful experimental design and data interpretation.
At the genome level, assays focus on confirming intended edits and identifying unintended off-target cleavage. Whole Genome Sequencing provides an unbiased assessment of modifications across the entire genome. However, detecting rare off-target events within a heterogeneous cell population requires immense sequencing depth, making this approach computationally intensive and financially burdensome. Standard bulk targeted sequencing offers a more focused and highly sensitive method for detecting localized indels at predicted sites. Yet, this approach generates a population average that completely masks zygosity and obscures the co-occurrence of on-target and off-target edits within individual cells. Furthermore, specialized cleavage assays like GUIDE-seq or CIRCLE-seq can map potential double-strand breaks but are technically complex and frequently suffer from high background noise.

At the transcriptome level, CRISPR analysis evaluates how genome editing alters gene expression. One method is called Perturb-seq which integrates pooled CRISPR screening with single-cell RNA sequencing. By utilizing expressed guide barcodes or direct guide RNA capture methods, Perturb-seq associates a specific genetic perturbation with whole-transcriptome expression profiles within individual cells. While Perturb-seq is highly informative, it assigns transcriptional readouts to guide identity rather than confirming the exact CRISPR gene editing outcomes, leaving a critical gap between intended perturbation and actual genomic results such as zygosity and co-occurrence, which are critical for understanding the efficacy and safety of CRISPR gene editing. Beyond Perturb-seq, developers frequently deploy standard bulk RNA sequencing to assess global transcriptional shifts, targeted RNA sequencing for sensitive analysis of specific functional pathways, or alternative implementations of the same single-cell CRISPR screening approach, including CROP-seq and CRISP-seq. However, many of these methods produce population-averaged data that entirely masks the crucial transcriptional heterogeneity existing between successfully edited, unedited, and off-target cells within a sample population. Furthermore, similar to Perturb-seq, these alternative single-cell transcriptomic approaches inherently link the expression profile to guide identity rather than a confirmed gene editing modification. This methodology assumes a successful edit occurred but fundamentally fails to verify the actual physical edit. Ultimately, relying exclusively on these transcriptomic modalities leaves a profound analytical void, preventing the definitive correlation of an observed transcriptional readout to a verified genotype within the exact same cell following a CRISPR experiment.
At the proteome level, evaluating the functional endpoint of a gene edit requires quantifying protein expression within the context of the edited cell. Flow cytometry remains one of the most widely adopted methods for protein quantification, enabling high-throughput measurement of surface and intracellular markers across large cell populations. However, conventional flow cytometry is inherently limited in multiplexing capacity and provides no genetic context; protein expression is measured with no linkage to the underlying genotype of individual cells. Other methods include Indexing of Transcriptomes and Epitopes by sequencing (CITE-seq) and RNA Expression and Protein sequencing (REAP-seq) advance resolution by using oligonucleotide-conjugated antibodies to detect specific proteins concurrently with transcriptomic profiling at the single-cell level. However, these approaches are inherently constrained by antibody availability and specificity. Across all of these modalities, a fundamental gap persists is that they do not have the ability to directly link a protein-level change to a confirmed CRISPR genome edit within the same individual cell.
| Assay | Attribute(s) it analyzes | Limitation | Solution |
|---|---|---|---|
Whole Genome Sequencing (WGS) | Unbiased genomic assessment of intended edits and unintended off-target cleavage events across the entire genome. | As a bulk method, WGS averages signals across the entire cell population, masking zygosity, edit co-occurrence, and cellular mosaicism. Additionally, detecting rare off-target events requires immense sequencing depth, making it computationally intensive and financially burdensome. | Tapestri provides targeted, highly sensitive single-cell DNA sequencing to resolve rare subclonal mutations and off-target events at the individual cell level without the massive sequencing depth requirements of bulk WGS. |
Cleavage Assays (e.g., GUIDE-seq, CIRCLE-seq) | Maps potential double-strand breaks and global off-target cleavage events. | Technically complex to execute and frequently suffer from high background noise, making definitive variant calling difficult without downstream phenotypic context. | Tapestri accurately confirms the presence of off-target edits and predicted chromosomal translocation junctions directly from the biological sample with high precision, linking them to specific cellular subpopulations. |
Perturb-seq, CROP-seq, CRISP-seq | Single-cell transcriptome profiling associated with specific genetic perturbations via expressed guide barcodes or direct gRNA capture. | Assigns transcriptional readouts to guide identity rather than a confirmed edit. It assumes a successful edit occurred but fundamentally fails to verify the actual physical genomic modification. Ideal for primary screens. | Tapestri’s two-step microfluidic workflow simultaneously captures targeted genomic loci and specific mRNA transcripts within the exact same cell, providing definitive genotype to transcriptome linkage rather than relying on guide RNA inference. Ideal for secondary and tertiary screens. |
Flow Cytometry | High-throughput functional quantification of cell surface and intracellular protein expression markers. | Inherently limited in multiplexing capacity and completely lacks genetic context, providing no linkage between the observed protein expression and the underlying genomic edit. | Tapestri utilizes custom oligonucleotide-conjugated antibodies alongside targeted DNA sequencing to confirm genomic edit with its corresponding immunophenotypic shift within the exact same individual cell. |
CITE-seq, REAP-seq | Concurrent transcriptomic profiling and surface protein detection (via oligonucleotide-conjugated antibodies) at the single-cell level. | Constrained by antibody availability and specificity. Crucially, these methods lack DNA-level resolution, failing to directly link the observed multiomic readouts to a physically confirmed CRISPR genome edit. | Tapestri consolidates targeted DNA, RNA, and protein measurements into a single, unified workflow, establishing the determinative genotype-to-phenotype evidence necessary for rigorous preclinical and clinical characterization. |
Using Single-Cell Multiomics To Assess CRISPR Genomic Edits
Single-cell multiomics is a critical methodology for CRISPR gene editing analysis, enabling the simultaneous co-detection of DNA, RNA, and protein attributes from the exact same individual cell. This approach directly resolves the analytical and QC blind spots associated with standard bulk approaches, replacing ambiguous inferences with the definitive, cell-by-cell data required for rigorous cell & gene therapy product characterization and gene editing experiment.
Utilizing specialized microfluidic platforms like Mission Bio's Tapestri, individual cells are encapsulated into nanoliter-volume aqueous droplets surrounded by an oil emulsion. A defining feature of the Tapestri platform is its two-step microfluidic workflow, which is absolutely essential for accessing tightly packaged genomic DNA. In the first step, encapsulated cells are lysed and treated with proteases to digest histone proteins and expose the chromatin. In the second step, these droplets are merged with barcoded beads and amplification reagents. To detect multiple attributes simultaneously, the platform supports distinct workflow configurations depending on the molecular modalities being interrogated. For genomic and transcriptomic co-detection, target-specific endogenous cellular RNA and targeted genomic loci are captured within the same droplet and tagged with an identical cell-specific barcode, enabling direct linkage of genotype to transcriptional state at single-cell resolution. For genomic and proteomic co-detection, cells are pre-incubated with targeted antibodies conjugated to DNA oligonucleotides prior to encapsulation. During the two-step droplet process, the exposed genomic DNA and the antibody-derived oligonucleotides are simultaneously barcoded and amplified via targeted multiplex polymerase chain reaction, preserving genomic target integrity while capturing the exact immunophenotype of each individual cell.
Single-cell multiomics isolates and quantifies distinct cellular attributes simultaneously (Figure 1). By retaining strict cellular resolution, single-cell multiomics accurately measures zygosity, identifies rare mutation co-occurrences, and resolves the full spectrum of editing outcomes from complete biallelic knockouts to heterozygous or mosaic edits at the level of the individual cell. On-target efficiency is confirmed while off-target mutations are identified and directly linked to aberrant transcriptomic or immunophenotypic signatures within isolated subclones. Chromosomal aberrations, copy number variation, and vector copy number are simultaneously assessed, providing a comprehensive view of genome integrity and construct integration status across the edited cell population. Biodistribution analysis further resolves which cell subsets harbor edits and in what proportion within tissue samples. Critically, all of these genotypic observations are linked to their downstream transcriptional and immunophenotypic states within the same cell enabling the definitive genotype-to-phenotype correlation essential for understanding therapeutic response needed in regulatory submissions and generating the cell-level evidence desired for manuscript and grant proposals.
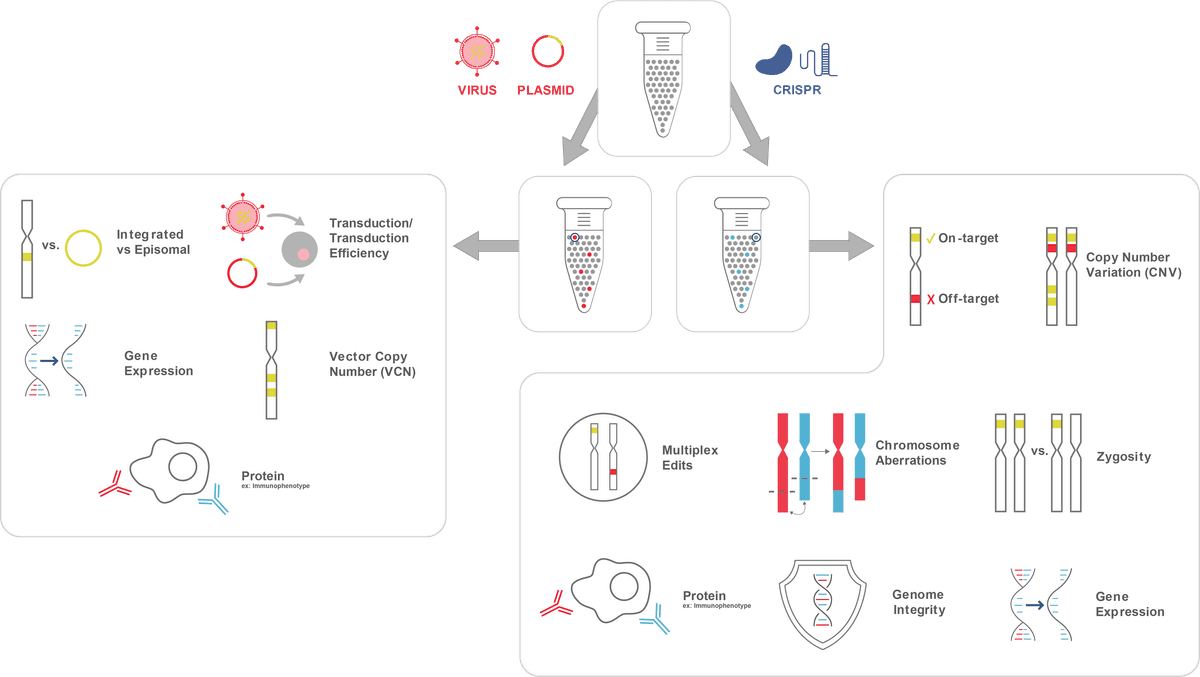
Figure 1: Possible attributes to considering evaluating following CRISPR gene editing.
Mission Bio Gene Editing and Genome Integrity Solutions
Mission Bio's Tapestri platform is a highly specialized single-cell multiomics instrument engineered specifically for resolving complex genomic heterogeneity and advancing CRISPR gene editing analysis. The Tapestri Genome Editing Solution offers a streamlined, automated bioinformatics pipeline designed specifically to bypass the computational limitations of many of the methods described above. It generates comprehensive metrics crucial for validating CRISPR gene editing efficacy and safety. The platform unambiguously resolves whether edits are monoallelic or biallelic and accurately quantifies the exact co-occurrence of multiplexed gene edits within individual cells. Crucially, the solution accurately confirms the presence of predicted chromosomal translocation junctions formed between on-target and off-target editing sites, providing vital safety data without requiring extensive, time-consuming clonal outgrowth. By incorporating custom oligonucleotide-conjugated protein panels, the solution directly correlates a specific genetic knockout with the corresponding functional loss of cell-surface protein expression.
To directly link confirmed genomic edits to their downstream transcriptional consequences within the same cell, the Tapestri Genome Editing Solution utilizes simultaneous targeted genotype to gene expression profiling. This multiomics capability enables researchers to confirm that specific on-target CRISPR modifications successfully drive the expected transcriptional knockdown or activation. Furthermore, it identifies discordant subpopulations where an edit is confirmed at the DNA level but the anticipated gene expression change is absent or aberrant. This level of resolution precisely distinguishes true functional knockouts from cells that retain residual transcript expression due to incomplete nonsense-mediated decay or compensatory transcriptional mechanisms. By co-measuring genotype and targeted RNA output within individual cells, the platform provides a direct, mechanistically grounded validation of editing efficacy that genomic confirmation alone cannot deliver.
Because chromosomal instability is a primary safety concern following a CRISPR gene editing event, Mission Bio developed the Tapestri Genome Integrity Solution. This solution leverages a highly uniform, five-hundred-amplicon panel to conduct whole-genome copy number variation analysis at single-cell resolution. This allows researchers to detect subclonal aneuploidy, chromosomal arm-level gains and losses, and copy-neutral loss of heterozygosity, which are frequently missed by standard bulk assays. By directly integrating copy number variation data with highly specific point mutations, targeted gene expression profiles, and immunophenotyping, the Tapestri platform delivers definitive insights into into genome stability, tumor evolution pathways, and the underlying mechanisms of therapeutic resistance consolidating what would otherwise require multiple orthogonal assays into a single, high-resolution workflow.
Single-Cell Services For Cell & Gene Therapy Development
Other CRISPR applications include Cell & Gene Therapies. Transitioning a CRISPR-edited therapy from preclinical development to clinical manufacturing requires rigorous analytical validation that standard methodologies consistently struggle to support. To address the intensifying regulatory scrutiny on product characterization, Mission Bio offers specialized Cell and Gene Therapy Assay Development Services. This dedicated team partners with therapeutic developers to design, optimize, and qualify custom single-cell multiomic assays tailored specifically to the unique therapy pipeline needs. By translating the multiparametric capabilities of the Tapestri platform into robust, GxP-deployable workflows, this service provides the exact analytical framework required to satisfy evolving Chemistry, Manufacturing, and Controls (CMC) requirements for cell and gene therapy development.
The fundamental value of this service lies in the strategic consolidation of complex quality control matrices into a single, fit-for-purpose multi-attribute analysis. Rather than relying on a fragmented suite of orthogonal assays to measure individual Critical Quality Attributes (CQAs), therapeutic developers can utilize a unified Tapestri workflow to simultaneously quantify vector copy number (VCN), transduction heterogeneity, biodistribution, and multiplex edit co-occurrence at the individual cell level (Figure 1). The team rigorously optimizes these custom assays to establish highly sensitive Lower Limits of Detection (LLOD), ensuring the reliable resolution of rare off-target activity, chromosomal aberrations, and sub-clonal drift. This consolidated approach generates the support, regulatory-relevant datasets necessary to prove CQA uniformity, demonstrate precise manufacturing comparability across scaling or reagent changes, and provide robust biological justification for clinical release specifications. Mission Bio’s Cell and Gene Therapy Assay Development Services systematically de-risk Investigational New Drug (IND) and Biologics License Application (BLA) submissions. This enables developers to definitively satisfy stringent FDA safety and potency expectations while significantly accelerating their clinical approval timelines.

The ongoing maturation of CRISPR gene editing technologies has necessitated a fundamental shift in analytical and bioinformatic methodologies. Traditional assays are proving insufficient for managing the nuanced technical challenges of mosaicism, variant zygosity, allele dropout, and widespread structural variations. The transition to single-cell multiomics elegantly resolves these critical limitations by maintaining the precise molecular integrity of individual cells. By simultaneously analyzing the genome, transcriptome, and proteome, advanced platforms like Tapestri allow researchers to establish direct, unambiguous links between a targeted CRISPR gene edit and its downstream expression and cellular phenotype. As CRISPR applications continue to mature across areas like disease modeling, functional genomics and screening, and cell and gene therapy, adopting single-cell multiomics is absolutely essential enabling researchers and developers alike to move from guide-level assumptions to confirmed, cell-level evidence that guarantees both the safety profiles and therapeutic efficacy of engineered genetic medicines.
If you want to learn more about how single-cell multiomic genome editing and genome integrity solutions can be used in your studies, contact us to learn more.
